FAQ page under construction (please send questions!) |
| How is a paired-alignment generated? |
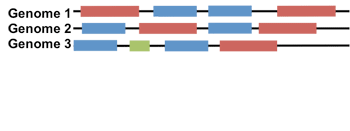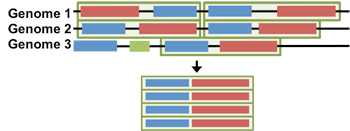 |
Lets say we want to build a paired-alignment between the BLUE gene and the RED gene. The task is further complicated by the fact that each genome has multiple copies of BLUE and RED (these are called paralogs). How do we know which copy of BLUE to pair with which copy of RED?
It turns out, for bacterial genomes, interacting genes are often found near eachother in the genome, in units called operons. Using the operon information, or a measure of how far apart two genes are in the genome (Δgene) we can avoid the paralog issue altogether!
Click here for technical details
|
- For the paper we exploited the fact that most UniProtKB/TrEMBL accession id(s) were assigned sequentially for each genome, allowing the rapid calulation of Δgene by simply taking the aphanumeric difference of the uniprot id(s). For the web-server, we use a lookup table of each uniprot id to its respective genome/contig and the nucleotide span, to calculate the Δgene.
- Download: uni2loc_2015_06_r124.raw.gz
- The mapping between uniprot id(s) and CDS records as extracted from the ENA database. The CDS record tells you the contig accession id and the location on that nucleotide sequence. A contig is continuous sequence of DNA.
- Will be updated following the ENA update cycle.
- Download: uni2loc_2015_06_r124.gz
- A processed mapping limited to uniprot id(s) that are found in multi-gene contigs.
- The list is sorted by uniprot id, allowing for use with sgrep for quick look-up.
- Lets say you are trying to decide if these two sequences should be stitched from two seperate alignments:
B3MJ72 07aQ_00I9_F (from alignment A)
B3MJ73 07aQ_00Ia_F (from alignment B)
The "07aQ" tells you they are from the same genome/contig.
The Δgene = |00I9-00Ia| = 1; tells you they are one unit apart (Note: gene count is in base-62 (0-9,a-z,A-Z)]).
The fact that both have "F" (which can be R), tells you that the protein-coding genes are running in the same direction, and thus more likely to be part of the same operon (if Δgene is small).
- Download: uni2loc_2015_06_r124.fas.gz
- Uniprot sequence database, filtered to remove singletons (contigs with only a single gene).
- The fasta headers:
>B3MJ72_07aQ_00I9_F
>B3MJ72_07aQ_00I9_F
- The advantage of using a pre-annotated database, is that you can quickly stitch the alignments in the future!
- Download: scripts_21Sep2018.zip (various helper scripts)
- stitch.pl - v1.4 bug fixed to deal with the new longer uniprot id(s) in jackhmmer
- stitch.pl - v1.5 bug fixed to deal with the new longer uniprot id(s) in hhblits
- stitch.pl - v1.6 major bug fixed to deal with the new longer uniprot id(s)
- Suggested protocol for HHblits
===================================================
- WARNING - HHblits has a hard-coded limit of 65535 sequences.
To overcome this, you must modify the code as follows:
- in: src/hhdecl.C: EXTERN const int MAXSEQ=262140;
- in: src/hhfullalignment.C: long unsigned int lq[MAXSEQ];
- in: src/hhfullalignment.C: long unsigned int lt[MAXSEQ];
===================================================
Input sequences: A.fasta and B.fasta
hhblits -alt 1 -i A.fasta -oa3m A.a3m -d DATABASE -n 8 -e 1E-20 -maxfilt 100000000 -neffmax 20 -nodiff -realign_max 10000000
hhblits -alt 1 -i B.fasta -oa3m B.a3m -d DATABASE -n 8 -e 1E-20 -maxfilt 100000000 -neffmax 20 -nodiff -realign_max 10000000
hhfilter -i A.a3m -o A_cov75.a3m -id 100 -cov 75
hhfilter -i B.a3m -o B_cov75.a3m -id 100 -cov 75
stitch.pl -strand -uni2loc -a A_cov75.a3m -b B_cov75.a3m -min 1 -max 20 -out AB_cov75.fas
hhfilter -i AB_cov75.fas -id 90 -o AB_id90cov75.fas
===================================================
Some notes for stitch.pl
===================================================
- Designed to work with HHblits and uniprot20 clustered database (see below).
- -min/-max control Δgene range you wish to include in the final alignment
- -uni2loc flag enables the uni2loc lookup table (you must modify the script to include absolute paths to sgrep and uni2loc table (see below))
- if -uni2loc flag is not present, difference in uniprot accession ids (as described in the paper) will be used, given the organism(ox=) definition is present in the header.
- optional -strand flag will enforce stitching only between genes in the same strand-ness
- WARNING - UniProt removed redundant sequence entries from it's database on April 1, 2015, meaning the newer uni2loc table will be missing these. If you wish to use an older uniprot20 clustered database, you must use an older uni2loc table.
- For uniprot20_2013_03, use uni2loc table: 2014_06
-
For uniprot20_2015_06, use uni2loc table: 2015_06
-
The new database requires the following fixes in the HHsuite source code to work:
- in: src/hhdecl.C, increase the limits for both "maxcol" and "maxres" to 40000.
- Suggested protocol for Jackhmmer
===================================================
Input sequences: A.fasta and B.fasta
jackhmmer --incE 1E-20 -E 1E-20 -N 8 -A A.sto A.fasta uni2loc_2015_06_r124.fas
jackhmmer --incE 1E-20 -E 1E-20 -N 8 -A B.sto B.fasta uni2loc_2015_06_r124.fas
sto2fas.pl A.sto > A.fas
sto2fas.pl B.sto > B.fas
hhfilter -i A.fas -o A_cov75.fas -id 100 -cov 75
hhfilter -i B.fas -o B_cov75.fas -id 100 -cov 75
stitch.pl -strand -uni2loc_fas -a A_cov75.fas -b B_cov75.fas -min 1 -max 20 -out AB_cov75.fas
hhfilter -i AB_cov75.fas -id 90 -o AB_id90cov75.fas
===================================================
Some notes:
===================================================
- Notice for jackhmmer, we are using the pre-annotated sequence database
- -uni2loc_fas flag for stitch.pl tells the script to use this pre-annotation found in both alignments
- the sto2fas.pl not only converts stockholm to fasta format, but also creates a master-slave alignment,
removing positions
not in the first query sequence.
|
| How does e-value play a role in seperating the paralogs from orthologs? |
| Alternativetly, one could adjust the e-value threshold until only one copy (for each gene) is detected per genome. The paralog ratio tells you how many copies per genome are detected for the given settings. Though sometimes its the combination of both approaches that produces the best pairing! Both Δgene and the e-value threshold can be adjusted on our web-server. |
| Is it possible to run GREMLIN on a complex that involves more than 2 genes? |
- Yes! You can do a all-vs-all paired analysis. For example if you have genes A,B and C. You can run the protocol on A+B, A+C, B+C (and put all the results together). This is what we did for the Ribosome and the NADH dehydrogenase complex!
|
| What is "S_sco" or "I_Prob"? |
- Scaled Score = raw_score/average(raw_scores)
- Referred to in the paper as "normalized coupling strength", see Figure 1 of our complexes paper to see how this score behaves with varying number of sequences.
- A coupling strength larger than one indicates higher than average coupling between two residues.
- I_Prob ≈ P(contact | scaled_score, seq/len, top_inter_score)
- Referred to in the paper as "GREMLIN score"
- If you do NOT know if the proteins interact, this score should help decide if there is enough information to infer an interaction. Otherwise you should use "Prob".
WARNING: The "I_Prob" score was trained on "obligate" interactions of the ribosome. Sometimes we find "transient" interactions to have a low prob (due to low relative coupling score), but still very accurate. Examples include the CH10 - CH60 (chaperonin) and DHSA - DHSB (Succinate dehydrogenase). We present all of our predictions to allow data-mining for more cases!
- Prob ≈ P(contact | scaled_score, seq/len)
- See FAQ for details about this score.
- The scores above should not be used as "cutoffs". For docking simulations we find that using ALL inter contacts within the top 1.5L intra/inter contacts as sigmoidal restraints to work best. "L" being the length of the protein-pair.
- Technical details of how to get these scores can be found in the question below!
|
| How do I run GREMLIN locally? |
- First you need an alignment
- (see technical details in the first question on how to generate this).
- Remove positions from the alignment that have > 75% gaps
- seq_len.pl -i AB_id90cov75.fas -percent 25
- AB_id90cov75.cut.fas - fasta file for your records
- AB_id90cov75.cut.msa - alignment to be used as input to GREMLIN
- AB_id90cov75.cut - mapping between the full length sequence and cut/trimmed sequence
- Please take note of seq_len value reported at the end of the script run, you'll need this value later!
- Run GREMLIN and get scores
- run_gremlin.sh MCR_location AB_idcov75.cut.msa AB.mtx MaxIter 30 verbose 1 apc 0
- AB.mtx - raw matrix from GREMLIN (apc 0 = no All Product Correction)
- mtx2sco.pl -mtx AB.mtx -cut AB_id90cov74.cut -div 100 -seq_len 5 -apcd AB.apcd
- AB.apcd - specialized All Product Correction to account for potential differing rates in each gene.
- -div 100 is the length of gene A (full length of gene A, not the cut length), aka the point of division in the matrix!
- -seq_len 5 is the number of sequences per length (this is the value you get at the end of running seq_len.pl)
- The scripts used in this demo: scripts_21Sep2018.zip. Use the download form to get a copy of GREMLIN.
|