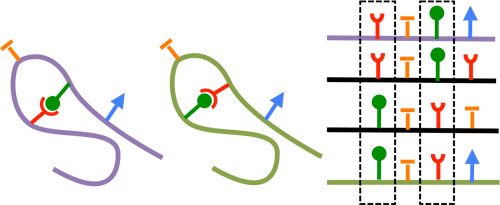
- For protein coding genes, when a residue mutates a compensatory mutation follows. These mutations are captured in our DNA and in the DNA of all living organisms. By analyzing a MSA (multiple sequence alignment) of homolgous protein sequences, we can measure coupling of any given residue pairs.
- Example on the left shows two shapes complementing eachother (red and green). If one of them changes, the other has to change. By comparying positions in a MSA, we can determine which pairs of positions might be in contact.
GREMLIN (Generative REgularized ModeLs of proteINs) is a method to learn a statistical model that simultaneously captures conservation and coevolution in a multiple sequence alignment. The statistical model, a Markov Random Field with log-linear potentials, has also been referred to as a maximum-entropy model or a global statistical model.
- GREMLIN's learning procedure optimizes a regularized Pseudo-likelihood objective resulting in a statistically consistent method. This results in higher accuracy than other approaches.
- For proteins with deep alignments, the strength of the co-evolution parameters after correcting for entropic effects with the Average Product Correction (APC) accurately predicts residue-residue contacts in the 3D structure of a protein.

- A contact map (or a distance matrix) is a two-dimensional representation of the three-dimensional protein structure showing which residues are in contact for a specified distance cutoff.
- Example on the left, shows a contact (yellow dashed line) between two strands of a protein structure. The same contact is also shown on the contact map as a yellow-filled circle. Notice the rainbow color on both the structure and contact map. The blue region is in contact with the green region.
- Just like we can display actual contacts for a given protein, we can also display covarying residues! See example here.
- For proteins that have no structure, the co-evolution measurements could be used as a predictor of which residues are in contacts.
- If a structure does exists, co-evolution measurements can be used to learn which contacts are actually important and evolutionarily selected for.
To make life easier we now provide a "Prob" which is probability of the residue pair being in contact, given the scaled_score and the number of sequences per length. Prob = P(contact | Scaled_score, seq/len).
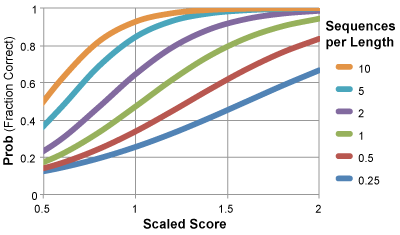
What is Scaled Score?
- Scaled Score is simply the raw score divided by the average of raw scores reported. If a value >> 1, this means that particular residue pair are highly covarying compared to the average.
- Scaled_score = (raw_score/average(raw_scores)).
- The value of the raw_score is the function of the learning procedure, L2 normalization and APC (entropic) correction.
How do you decide which top raw_scores to use?
- If we ignore the immediate neighbors (sequence seperation >= 3), we find that on average each residue makes 1.5 contacts. We thus select the top 1.5L contacts (L = length or protein after trimming to remove gappy poisitions) for our analysis.
- By non-redundant we mean that the no pair of sequences in the input alignment are more than 90% identical to each other.
- We use HHblits to find homologous sequences.
- HHblits and HHsearch(see question below) are part of the HHsuite package (that use Hmm-Hmm alignments, hence the "HH").
- HHblits works similar to PSI-Blast, but is much faster and more accurate. It achieves the speed in part by searching against a pre-clustered Uniprot database. Once a cluster is detected all the sequences from the cluster are dumped into the alignment. Given that the pre-clustering is very computationally expensive, it is only performed once a year.
- What is Jackhmmer?
- Jackhmmer is part of the HMMER package. It is similar to HHblist, but slower since it compares to all sequences (instead of clusters). Since pre-clustering it not required, it can use the latest Uniprot database directly which is updated once a month.
- The purpose of the HHsearch output is to give the user an idea of how well the GREMLIN output matches the contacts of the closest known PDB
structure. HHsearch results are not used in GREMLIN. - We use HHsearch to find homolgous PDB structures.
- For each PDB (entry in the Protein Database), a pre-computed alignment and corrresponding hmm (hidden markov model) is made.
- We then compare your query alignment (generated by HHblits) to the alignment of the HHsearch hit. If the sequences in the alignments are very different we report a HIGH HH_delta, otherwise the HH_delta is low.
- For homology modeling purposes, high HH_delta (>0.5) means that there is likely to be more information in the co-evolution matrix than if you were to simply copy all the coordinates of the closet known homolog.
- Assuming the same input alignment is used, the rank of the scores should be identical. The difference comes in how the scores are scaled. After the Average Product Correction (APC), standalone GREMLIN rescales the values to > 0. The web-server does not do this scaling and reports the APC values directly as the "raw score".
- The "raw scores" were meant to be used for ranking purposes only. The range of the value will vary depending on the length of the query sequence and the number of sequences.
- Yes, as long as you can create a paired alignment. Please refer to our help section below the Complex Submission form.
- Older algorithms such as MI (mutual information) were not very accurate for contact prediction because they are not able distinguish direct from indirect coupling. For example, if A is coupled to B and B to C, an analysis that only looks at pairs of interaction (local statistical model) will find that A is coupled to C. Newer algorithms use a global statiscal model which are better able distinguish direct from indirect coupling, by trying to find the most parsimonious coupling network. All the new methods (such as GREMLIN, DCA, PSICOV) agree on the model, but they differ in their learning procedure (and some underlying assumptions).
- We find that the Pseudo-likelihood learning procedure to be more accurate than DCA and PSICOV. The pseudo-likelihood method (first introduced in GREMLIN_v1) has been reimplemented as GREMLIN_v2 [what we use], plmDCA and most recently as CCMpred for contact prediction. DCA has been renamed to mfDCA (to distinguish it from plmDCA and other "DCA" variants) and most recently reimplemented as FreeContact.
- Evfold - now also uses Pseudo-likelihood learning procedure as default.
- MetaPSICOV - combines CCMpred, FreeContact and PSICOV.
- DCA - Direct Coupling Analysis (aka mfDCA)
- MISTIC - Mutual Information Server to Infer Coevolution
- CMAT - Correlated Mutation Analysis Tool, uses a corrected MI variant.
- PhyCMAP (RaptorX)
- MSAvolve - Matlab toolbox that includes MI, MIP, ZRES, ZPX, ZPX2, nb/db/dgb, 3D_MI, 4D_MI, DCA, plmDCA, hp_pca_DCA, PSICOV, GREMLIN, logR, OMES, McBASC, ELSC, SCA.
- CCMpred - open source implementation of GREMLIN/plmDCA in (C/CUDA C) for use with GPU/CPU and parallel computing.
- WARNING CCMpred (parallel implementation) can take up to 100 iterations to achive same accuracy as GREMLIN's 30 iterations, for large alignments.
With GREMLIN (serial implementation) we find the accuracy to not increase much after 30 iterations.
- WARNING CCMpred (parallel implementation) can take up to 100 iterations to achive same accuracy as GREMLIN's 30 iterations, for large alignments.
- FreeContact - open source implementation of EVfold-mfDCA/PSICOV in C++, which can be used as a python or perl module.
- PconsC - pipeline that combines PSICOV and plmDCA.
- See the list of some of the older programs compiled by Juan et al.
| - for other great tools check out OMICtools! |